 ВІСНИК ФАРМАКОЛОГІЇ ТА ФАРМАЦІЇ, № 10/2003
ВІСНИК ФАРМАКОЛОГІЇ ТА ФАРМАЦІЇ, № 10/2003
Леонид Громов, д-р мед. наук, профессор; Елена Дудко, канд. мед. наук
Институт фармакологии и токсикологии АМН Украины
Психофармакологии, как науке о веществах, действующих на головной мозг и изменяющих психические функции, исполнилось 50 лет. Ее рождение датируется 1952 годом, когда впервые был применен аминазин для лечения психических заболеваний. Это было начало первой группы психофармакологических средств — нейролептиков.
Затем были созданы новые психотропные препараты, что позволило классифицировать их следующим образом: нейролептики, транквилизаторы, антидепрессанты, психостимуляторы, ноотропы.
Нейролептики и транквилизаторы оказывают депримирующее влияние на центральную нервную систему, антидепрессанты, психостимуляторы и ноотропы — активирующее.
В каждую из этих групп психофармакологических средств, особенно, нейролептиков, транквилизаторов и антидепрессантов, входит довольно разнообразная номенклатура лекарственных препаратов, которые отличаются как по химической структуре, так и по механизму действия.
Во главе каждой группы стоят препараты с известным механизмом действия, получившие название «типичные».
Нейролептики — это производные фенотиазина и бутирофенона.
Антидепрессанты возглавляют ингибиторы МАО (моноаминоксидазы), а также избирательные и неизбирательные блокаторы обратного захвата моноаминов.
«Типичными представителями транквилизаторов являются производные 1,4-бензодиазепина. Разнообразная по химической структуре, фармакологической активности и основным элементам механизма действия группа психофармакологических средств, отличающихся от «типичных» нейролептиков, антидепрессантов и транквилизаторов, составляет соответственно «атипичные» нейролептики, антидепрессанты и транквилизаторы.
В данной работе рассматриваются «типичные» и «атипичные» транквилизаторы.
К «типичным» транквилизаторам, производным 1,4-бензодиазепина, относятся диазепам (сибазон), клоназепам, флуразепам, феназепам, лоразепам, альпразолам, оксазепам, медазепам, нитразепам, флунитразепам, триазолам, бротизолам, тетразе- пам, клобазам, гидазепам.
Все эти препараты в разной степени оказывают анксиолитическое, гипногенное, миорелаксирующее и транквилизирующее действие.
Основным положительным фармакологическим свойством «типичных» транквилизаторов является их анксиолитическое влияние, т.е. способность устранять тревогу, страх, панику, нервное напряжение.
В последние годы достигнуты большие успехи в понимании механизмов действия производных 1,4-бензодиазепина и небензодиазепиновых снотворных — зопиклона, золпидема. Это связано с расшифровкой молекулярного строения рецепторов ГАМК и ГАМК-бензодеазепинового рецепторного комплекса. Последний является биологической мишенью действия бензодиазепинов.
Фармакологически различают бикукуллинчувствительные ГАМКА-рецепторы и баклофенчувствительные ГАМКБ-рецепторы. ГАМКА-рецепторы связаны с ионными каналами клеточной мембраны для ионов С1» (хлора). ГАМКБ-рецепторы ассоциируются с G-протеин-связанными рецепторами и Са2+-каналами [9, 12].
Наиболее хорошо изучены ГАМКА-рецепторы. Установлено, что ГАМКА-рецептор — это пентамерный белок, состоящий из пяти самостоятельных протеинов, которые образуют розетку вокруг мембранного канала для ионов хлора. Открытие канала для трансмембранного тока ионов С1 внутрь клетки вызывает гиперполяризацию мембраны, что определяет ингибирующую (тормозную) функцию нейрона. Лигандом этих каналов является гамма аминомасляная кислота (ГАМК) [11, 16, 21]. На эти каналы действуют бензодиазепины и небензодиазепиновые снотворные [17]. Белки ГАМКА-рецепторов состоят из целого ряда субъединиц (а, р, у), которые в свою очередь подразделяются на подтипы (а1, Р13, у2 и т.д.) [8, 17]. Теоретически предполагается наличие 36 подтипов субъединиц ГАМКА-рецептора. Структурное образование субъединиц белков ГАМКА-рецептора закодировано в человеческой хромосоме 5 [22]. Каждый из белков подтипов субъединиц ГАМКА-рецепторов имеет якорные аминокислоты, с которыми связываются ГАМК-позитивные, ГАМК-негативные, бензодиазепиновые и снотворные вещества, что во многом детерминирует их фармакодинамический профиль действия (рис. 1). Считается, что на расщепленной поверхности ГАМКА-рецептора между, а и у его субъединицами встроен бензодиазепиновый рецептор, с которым взаимодействуют бензодиазепиновые транквилизаторы и снотворные средства, в том числе небензодиазепиновой структуры (зопиклон, золпидем) [12]. Эта молекулярная структура получила название ГАМК-хлорбензодиазепиновый
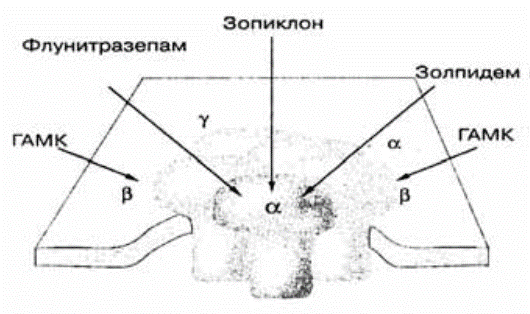
Рис. 1.Схематическое представление ГАМК-рецептора млекопитающих, встроенного в мембрану клетки; показаны а, в, и у-субъединицы и связывающие участки для ГАМК и снотворных средств.
рецепторный комплекс (рис. 2). Бензодиазепины связываются с якорными аминокислотами субъединиц ГАМК-хлорбензодиазепинового рецепторного комплекса внутри хлорного канала, тогда как снотворные средства — на поверхности этого канала. Ключевыми аминокислотами для а-субьединицы ГАМКА-рецептора являются гистидин (Hig) в положении 102 аминокислотной последовательности белка, тирозин (Туг) — 159 и глицин (Gly) — 200. Для у-субъединицы — фенилаланин
(Phe) -77, метионин (Met) — 130 и треонин (Thr) — 142 [7, 23]. Связывание с этими ключевыми аминокислотами в различных субьединицах ГАМК-хлорбензодиазепинового комплекса определяют физиологические и фармакологические эффекты эндогенных и экзогенных лигандов этого рецепторного комплекса (рис. 3).
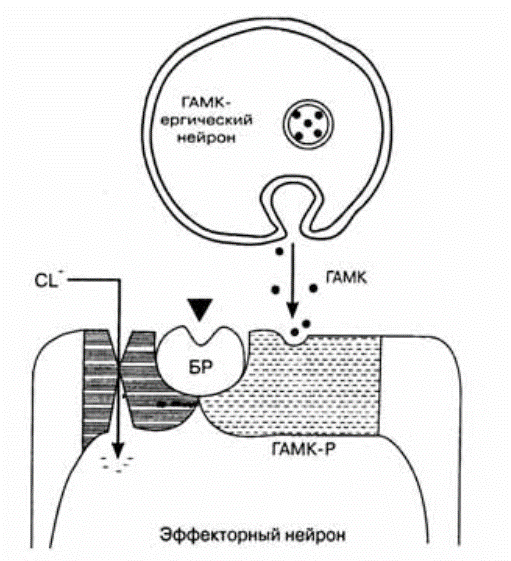
Рис. 2. Предполагаемый молекулярный механизм действия бензодиазепинов.
БР — бензодиазепиновый рецептор; бензодиазепиновая молекула изображена в виде треугольника; ГАМК-Р — ГАМК-рецептор;
С1 — хлорид, проходящий через хлорный канал.
При этом, в отсутствие ГАМК, бензодиазепины не влияют на хлорную проводимость нейрональных мембран. Более того, они не влияют на число хлорных каналов и движение ионов С1-, но удлиняют возможность существования открытых ионных каналов в ответ на действие ГАмК.
Среди нежелательных побочных эффектов бензодиазепинов наиболее существенными являются миорелаксация, снижение памяти и развитие физической и психической зависимости. Синдром зависимости формируется при хроническом лечении определенными бензодиазепинами [18].
Показано, что при длительном введении бензодиазепинов снижается количество РНК мессенджера, кодирующего различные субъединицы ГАМК- бензодиазепинового рецепторного комплекса в коре мозга. При этом снижается экспрессия генов а1, Р2, у2 субъединиц, которые являются самыми распространенными в ГАМКА-рецепторе. Это снижение регуляции транскрипции генов, которые кодируют образование субъединиц а1, Р2, у2, происходит на 5 хромосоме [13]. Такие же изменения (но более ограниченные) транскрипции генов субъединиц наблюдаются при длительном введении зопиклона и золпидема [14], что может приводить к развитию зависимости при хроническом применении этих снотворных средств.
К «атипичным» транквилизаторам относятся амизил, оксилидин, адаптол, ноофен, тофизопам, триоксазин, азапироновые производные (буспирон, ипсапирон, тандоспирон, гепирон). В связи с выраженной ю-холинолитической активностью амизил и оксилидин в настоящее время в клинической практике в качестве транквилизаторов практически не используется. Тофизопам, ноофен и триоксазин наряду с транквилизирующим действием оказывает определенное психостимулирующее влияние.
Группу «атипичных» транквилизаторов составляют различные по химической структуре и во многом с неизвестным механизмом действия лекарственные препараты, которые также оказывают анксиолитическое действие, но не вызывают миорелаксации, мнестических расстройств и синдрома зависимости.
Транквилизирующее влияние препаратов семейства буспирона определяется их способностью модулировать серотонинергическую передачу, которая наряду с ГАМК-ергической системой в ЦНС выполняет ингибирующую (тормозную) функцию.
Своеобразными психофармакологическими свойствами обладает Адаптол (2,4,6,8-тетраметил-2,4,6,8- тетраазибицикло (3,3,0) октандион-3,7). Этот препарат оказывает умеренное транквилизирующее влияние и практически не вызывает побочных эффектов. Адаптол по химическому строению близок к метаболитам организма, т.к. состоит из двух молекул мочевины. Поэтому считается, что препарат может оказывать метаболическое действие, нормализуя нарушенные стрессом различные метаболические процессы; кроме того, адаптол может влиять на функционирование нейромедиаторных систем. Однако механизм действия адаптола до конца не выяснен [5]. Согласно данным литературы, препарат проявляет антагонистическую активность по отношению к возбуждающей адренергической и глутаматергической системам и усиливает функционирование тормозных серотонин— и ГАМК-ергических механизмов мозга.
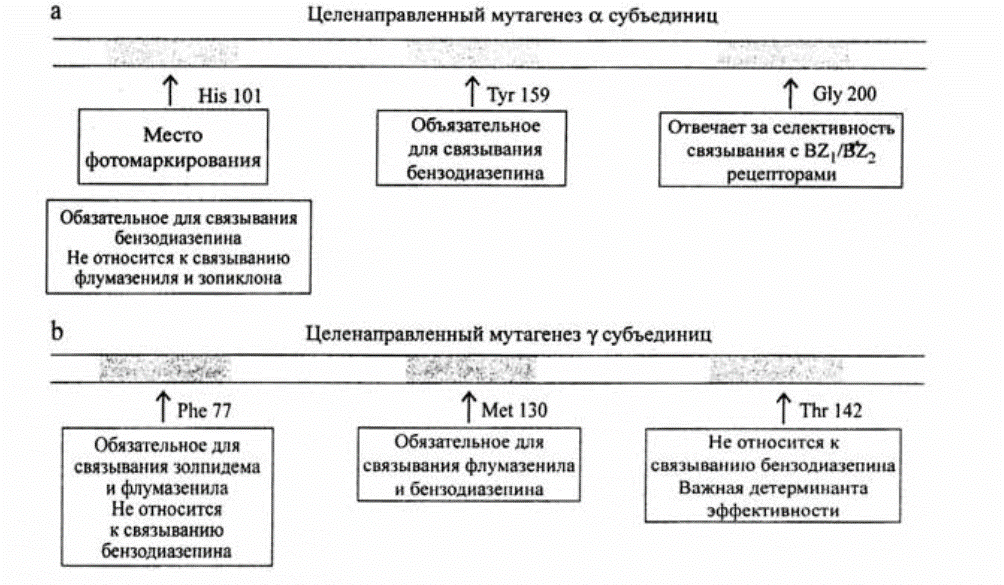
Рис. 3. Роль индивидуальных аминокислот ГЛМКА-рецептора в связывании с бензодиазепинами и другими препаратами. Показаны аминокислоты на (а) а (вверху) и (Ь) у (внизу) субъединицах ГЛМКА-рецептора, чьи мутации изменяют взаимодействия при связывании препарата. Предполагаемая роль каждой из аминокислот обозначена в рамках.
Такое представление обосновывается тем, что препарат ингибирует фенаминовое возбуждение, снижает уровень норадреналина в мозге и снижает токсичность фенамина, а также препятствует повышению содержания глутамата в мозге, вызываемому стрессом. Наряду с этим, адаптол повышает уровень серотонина в крови и в стволе мозга. В мозге повышается также количество ГАМК. Кроме того, адаптол усиливает ареколиновый тремор [5]. Следует, однако, отметить, что указанные эффекты регистрировались при введении больших доз адаптола (10001500-2000 мг/кг), что составляет 1/2-1/3 от ЛД50 если принять во внимание, что ЛД50 препарата находится в пределах 3500 мг/кг. Учитывая достаточную лечебную эффективность адаптола и его популярность среди врачей и пациентов, нами были предприняты систематические исследования, направленные на выяснение механизмов действия этого препарата. В наших исследованиях, при изучении механизма действия адаптола, был использован метод фармакологического анализа функционирования нейромедиаторных систем. С этой целью оценивалось влияние адаптола на основные эффекты селективных анализаторов функционирования ГАМК-, глицин-, глутамат-, адрено-, дофамино— и холинергической систем. Адаптол вводился внутрижелудочно в дозе 500 мг/кг за 1 ч до внутрибрюшинной инъекции соответствующих анализаторов (стрихнин- глицинергическая система; пикротоксин, тиосемикар- базид, бикукуллин,коразол-ГАМК-ергическаясистема; никотин, ареколин, эзерин-холинергическая система; апоморфин, галоперидол-дофаминергическая система, сиднокарб-адренергическая система; каиновая кислота-глутаматергическая система). Проведенные исследования показали, что адаптол не предупреждал развитие судорог при введении коразола, пикротоксина, бикукуллина, тиосемикарбазида, стрихнина, каиновой кислоты и никотина. Следовательно, адаптол не оказывает существенного влияния на ГАМК-, глицин-, глутамат— и Н-холинореактивные системы. Препарат также не изменял эффекты ареколина и эзерина. Наряду с этим, адаптол снижал выраженность галоперидоловой каталепсии, но не устранял апоморфиновую стереотипию. Препарат предупреждал сиднокарбовую гипертермию, но усиливал двигательную гиперактивность, вызванную сиднокарбом. Эти результаты дают основания предположить, что адаптол, в плане действия на нейромедиаторные системы, оказывает основное влияние на катехоламинергическую систему, которая, как известно, является одной из ведущих нейрохимических систем реагирования при стрессорных воздействиях и эмоциональных реакциях. При этом можно констатировать, что адаптол сочетает свойства дофаминпозитивного и своеобразного агониста- антагониста адренергической системы. Это важно для понимания клинических эффектов адаптола, так как препараты с таким механизмом действия могут снижать явления страха, тревоги, эмоционального напряжения и одновременно с этим активировать нейрофизиологические функции при астеноневротическом синдроме.
Стрессорные воздействия инициируют развитие «оксидантного стресса». Активация при этом процессов свободнорадикального окисления является причиой структурнофункциональной дезорганизации клеточной мембраны, а значит, синаптической передачи.
В литературе имеются сведения о том, что адаптол оказывает прооксидантное действие [5]. В то же время, мочевина рассматривается как эталонный антиоксидант, наряду с а-токоферолом, дибунолом и другими референтными антиоксидантными препаратами [1,2,20]. Выше указывалось, что по химической структуре адаптол состоит из двух молекул мочевины. Все это оправдывало исследование возможной антиоксидантной активности адаптола.
Антиоксидантные свойства адаптола изучали по ингибированию супероксидрадикала в системе аутоокисления адреналина, по торможению процессов пероксидации в гомогенате ткани головного мозга, вызванных токсической концентрацией донаторов нитратов (нитрозирующий стресс) и по торможению окислительной модификации белков в условиях окислительного стресса in vitro [3, 10, 15]. Влияние адаптола на процессы свободно-радикального окисления (СРО) определялось по его антирадикальной активности (АРА), антиоксидантной активности (АОА) и содержанию малонового диальдегида (МДА). Полученные результаты приведены в таблицах 1-4.
Как видно из представленных данных, у адаптола обнаружен антиоксидантный эффект. При этом адаптол в концентрации в 1,5 раза ниже, чем мочевина, в 1,5 раза превышает мочевину по АРА (табл. 1). Данный факт свидетельствует о том, что находящиеся в структуре молекулы адаптола два остатка мочевины сообщают последней свойства «ловушки» супероксидрадикала. Этот эффект у адаптола более сильный, чем у мочевины.
Антиоксидантная активность адаптола, при неферментативном инициировании СРО, оказалось в 1,5 раза выше, чему известного антиоксиданта дибу- нола (ионола) (табл. 2). Адаптол проявляет также АОА в условиях моделирования нитрозирующего стресса in vitro, превышая действие дибунола в 2 раза (табл. 3) Подобный эффект адаптола связан с его способностью ингибировать супероксидрадикал (табл. 1) и, возможно, предотвращать образование ONOO-.
Исследование АОА адаптола в условиях окислительного стресса in vitro показало, что адаптол тормозит образование альдегидных продуктов окисления белков головного мозга крыс на 27,3% (при максимуме поглощения 270 нм) и карбоксильных (конечных) продуктов окисления белков (при 363 нм) на 34% (табл. 4). По этому тесту адаптол превосходит мочевину, которая вообще не тормозит образование карбоксильных продуктов окисления белков головного мозга крыс.
Таким образом, в механизме действия препарата «Адаптол» присутствует прямой антиоксидантный эффект, который заключается в способности ингибировать активные формы кислорода и за счет этого тормозить переоксидацию не только липидов, но и белков. Учитывая, что липидный бислой и белки составляют структурно-функциональную основу клеточной оболочки, которая нарушается при оксидантном стрессе, становится понятным ключевое значение антиоксидантных свойств адаптола в механизме его мембраностабилизирующего, адаптогенного и транквилизирующего действия.
Таблица 1
Антирадикальная активность адаптола по ингибированию супероксирадикала in vitro
|
Препарат |
Концентрация, мкМ |
оптическая плотность, д |
ара,% |
|
Контроль (адреналин) |
2,25-10-3 |
0,17±0,005 |
— |
|
Адаптол |
0,30 |
0,095±0,0015** |
44,0 |
|
Мочевина |
0,50 |
0,12±0,002** |
30,0 |
Примечание: в таблицах 1-4
* — достоверные различия по сравнению с интактной;
** — достоверные различия по сравнению с контролем.
Таблица 2
Антиоксидантная активность адаптола при неферментативном инициировании СРО in vitro
|
Препарат |
Концентрация, мкМ |
МдА, мкМ/мл |
APA,% |
|
Интактная |
— |
0,28±0,007 |
— |
|
Контроль (FeS04) |
25,0 |
4,50±0,07* |
|
|
Адаптол |
2,4 |
2,50±0,03** |
44,0 |
|
Дибунол |
3,0 |
3,2±0,04** |
28,0 |
Таблица 3
Влияние адаптола на содержание малонового диальдегида (МДА) в гомогенате головного мозга крыс при моделировании нитрозирующего стресса in vitro
|
исследуемые серии |
Концентрация, мкМ |
МдА, мкМ/г ткани |
% снижения |
|
Интактная |
— |
0,55±0,002 |
— |
|
Контрольная (стресс) Нитропруссид натрия |
— |
2,52±0,075* |
— |
|
Адаптол |
1,24 |
1,95±0,065** |
-22,6 |
|
Дибунол |
2,28 |
2,24±0,027* |
-11,0 |
Таблица 4
Влияние адаптола на окислительную модификацию белков в тканях мозга крыс при моделировании окислительного стресса in vitro
|
исследуемые серии |
Концентрация, мкМ |
Продукты окислительной модификации белка д.е. 1 г ткани |
|
|
270 им |
363 нм |
||
|
Интактная |
— |
4,08±0,08 |
12,6±0,10 |
|
Контрольная (стресс) Н202 - 50 мМ, FeS04 - 10 мМ |
— |
27,6±0,074* |
48,5±2,60* |
|
Адаптол |
1,24 |
20,1±0,52** |
32,0±1,14** |
|
Дибунол |
3,0 |
24,0±0,44 |
44,0±2,31 |
В пользу мембранотропного действия адаптола свидетельствуют еще ряд положений.
Первое. Квантово-химические расчеты электронных орбиталей молекулы адаптола и молекул аминокислот, составляющих белковый остов а— и у-субъединиц ГАМК-рецептора показали возможность средней силы взаимодействия препарата с треонином (Тhr 142) у-субьединицы (см. рис. 3). Эта аминокислота у-субъединицы не связывается с бензодиазепиновыми транквилизаторами.
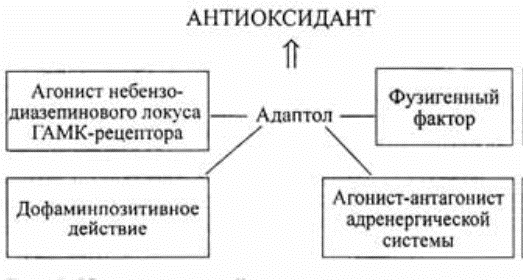
Рис.4. Механизм действия адаптола
Представленный анализ позволяет сделать заключение о том, что адаптол может быть агонистом ГАМКА-рецептора (небензодиазепинового его локуса) на пост— или пресинаптической мембране.
Второе. Из данных литературы [6] известно, что мочевина обладает сильным фузигенным действием, т.е. усиливает слияние мембран пресинаптических везикул с пресинаптической мембраной. Этот процесс инициирует зкзоцитоз, т.е. высвобождение медиаторов из пресинаптического депо.
Так как молекула адаптола состоит из 2 фрагментов мочевины, то можно предполагать, что препарат также обладает фузигенными свойствами, усиливая высвобождение тормозных и активирующих медиаторов, оптимизируя тем самым процессы возбуждения и торможения в центральной нервной системе.
На основании полученных результатов исследований и данных литературы можно представить схему механизмов действия адаптола (рис. 4).
Исходя из полученных экспериментальных данных, можно сделать вывод о том, что ключевым моментом в механизме действия адаптола являются его антиоксидантные свойства. Адаптол также демонстрирует свойства агониста-антагониста адренергической системы, что объясняет его выраженные нормостенические эффекты.
Кроме того, препарат обладает дофаминпозитивным влиянием, что клинически проявляется в его активирующем компоненте действия. Имеются экспериментальные предпосылки, свидетельствующие о том, что адаптол обладает фузигенной активностью и проявляет свойства агониста небензодиазепинового локуса ГАМК-рецептора. Таким образом, сочетание нейрометаболического и нейромедиаторного действия адаптола объясняет полифункциональность его нейрофармакологических эффектов.
Согласно определению Г.ААвруцкого (цит. по М.Д.Машковскому [4]), транквилизаторы, адресуясь главным образом к психопатологическим расстройствам невротического уровня, способствуют устранению широкого круга невротических и неврозоподобных расстройств, уменьшая, прежде всего, эмоциональную напряженность, тревогу и страх.
Анализ экспериментальных и клинических данных по механизму действия и фармакологическим эффектам «типичных» и «атипичных» транквилизаторов позволяет предложить схему выбора лечебной тактики фармакотерапии различной клинической симптоматики при неврозах и неврозоподобных состояниях.
На рис. 5 приведены варианты выбора «стандартизированной схемы лечения», в соответствии с современными воззрениями.
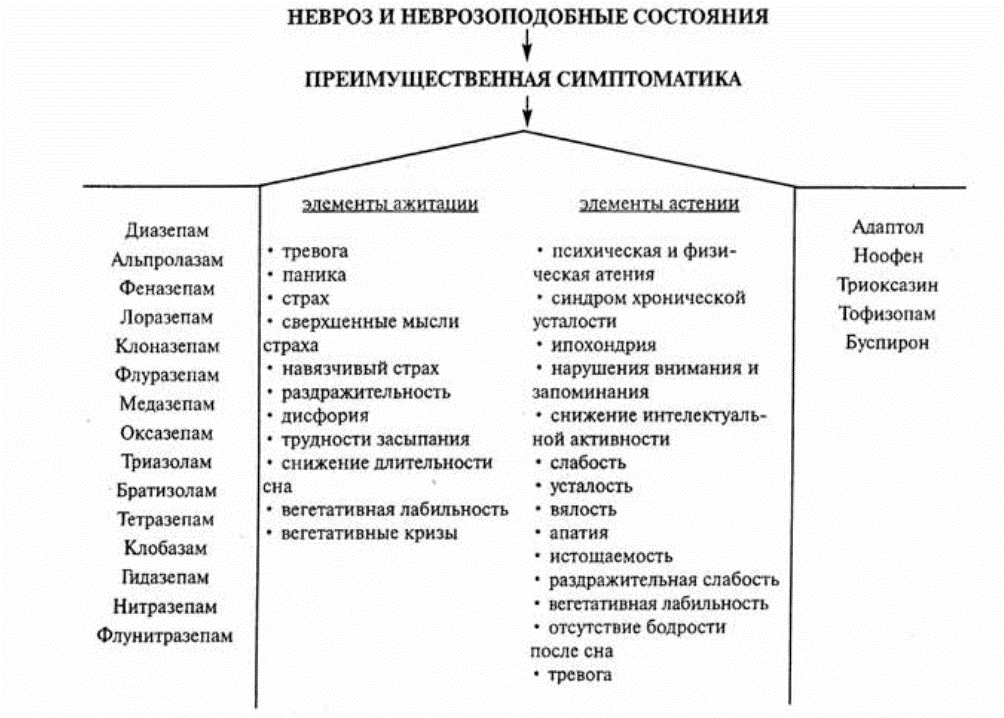
Рис. 5. Выбор стандартизированного лечения невротических и неврозоподобных состояний
ЛИТЕРАТУРА
- Биленко М.В. Ишемические и реперфузионные повреждения органов. — М.: Медицина, 1989. -368 с.
- Дунаев В.В., Беленичев И.Ф., Коваленко СИ. Антирадикальная и антиокислительная активность соединений призводных 1,2,4-триазола и хиназолина при ишемии мозга//Укр. биохим. журн. — 1996. — Т68. №1. — С. 100-104.
- Губский Ю.І., Дунаев В.В. Беленічев та ін. Методи оцінки антиоксидантних властивостей фізіологічно активних сполук при ініціюванні вільно радикальних процесів у дослідах in vitro: Метод. рекомендації. — К.: ДФЦ МОЗ України, 2002.-26 с.
- Машковский М.Д, Лекарственные средства. — М: Медицина, 1993. — Т1. — С. 86.
- Заиконникова И.В., Зимакова И.Е., Лебедев О.В., Хмельницкий Л.И. Мебикар. — М., 1990, — 45 с.
- Трикаш И.О., Терлецкая Я.Т, Колчинская Л.И., Малышева М.К. Способность латротоксинподобного белка головного мозга вызывать слияние отрицательно заряженых липосом//Нейрофизиол. — 1993. — №1 (5). — С. 329-334.
- Arin J. , Brooks-Kagal А. , Weiss О. S. Two tyrosine residues on the a subunit are crucical for benzodiazepine binding and allosteric modulation of aminobutyric acidA receptors/ /Мої. Pharmacol. — 1997. -V.51. — P. 833-841.
- Barnard E. A., Skolnik P, Bateson A.N.,Sieghart W International Unipn of Pharmacology-XV -Subtypes of g-aminobutyric acidA receptors — classification the basis of subunit structure and receptor function//Pharmacol. Rev. — 1998. — V50. — P 291-313.
- Chebib M., Johnston G.A.R. GABA-Activated Ligand Gated Ion Channels: Medicinal Chemistry and Molecular Biology//J. Med. Chem. - 2000. - V.43,№8. - P 1427-1447.
- Daneshvar В., Frandes H.,Autrup H. g-Glytamyl semialdehyde and 2-amino-adipic semialdehyde: Biomarkers of oxidative damarge to protein// Biomar kers. — 1997. — V.14, №2. — P 236-245.
- DevisPA^^nm M.C,Hales TG.,Kirkness E.F. Insensitivity to anaesthetic agents conferred by a class of GABAa receptor subtunit//Nature. - 1997. - V.385. - P 820-823.
- Doble A. New insights into the mechanism of action of hypnotics//J.Psychopharmacol. - 1999. - V13, №4 (Suppl.1). - P 11-20.
- Holt R.A, Batesoл A.N., Martin I.L. Chronic treatment with diazepam or abecamil differentially affects the expression of GABAa receptor subunit mRNAs in the rat cortex//Neuropharmacol. - 1996. - V.35. - P 1457-1463.
- Holt R.A.,Bateson A.N.,Martin I.L Chronic Zolpidem treatment alters GABAa receptor mRNA levels in the rat cortex//Eur. S-Pharmacol. - 1997. - V.329. - P 129-132.
- Hausladen A. NO and metalloprotein//Eur. Cell Biol. - 1998. - V. 75 (Suppl.48). - P 32-38.
- Johnston G.A. R. GABAa receptor pharmacology //Pharmacol. Ther. - 1996. - V.69. - P 176-198.
- Korpi E. R., Muttila M.J., Wisden W, Luddens H. GABAA receptor subtypes - clinical efficacy and selectivity of benzodiazepine site ligands // Ann. Med. - 1997. - V.29. - P 275-282.
- Ladez M. Benzodiazepines. A risk - benefit profit И CNS Drugs. - 1994. - V. 1. - P .377-387.
- Mehta A.K., Ticku M.K. An update on GABAa receptors//Brain Res. Rev. - 1999. - V.29. - P 196-217.
- Riondel J.,Glise D.,Fernandez-Carlos T. In Vitro comparative study of cytolysis mediated by natural Killes alls towards malignant alls preincubated with antioxidants//AnticancerRes. - 1998. - V.18, №3. - P 1757-1763.
- SieghartW Structure and pharmacology ofg-aminobutyric acidA receptor subtypes//Pharmacol. Rev. - 1995. - V47. - P 181-234.
- Whiting P.S., Mc. Kernan R.M., Wafford K.A. Structure and pharmacology of vertebrate GABAa receptor subtypes//Intl. Rev. Neurobiol. - 1995. - V.38. - P 95-138.
- Wieland H.A., Luddens H.,Seeburg PH. A single histidine in GABAa receptors is essential for benzodiazepine agonist binding// Biol. Chem. -1992. - V.267. - P 1426-1429.
